Tahoe-100M: A Giga-Scale Single-Cell Perturbation Atlas for Context-Dependent Gene Function and Cellular Modeling

作者:
Jesse Zhang*,1, Airol A Ubas*,1, Richard de Borja*,1, Valentine Svensson*,1, Nicole Thomas*,1, Neha Thakar **,1, Ian
Lai ,1, Aidan Winters,1, Umair Khan**,1, Matthew G
期刊:bioRxiv
重要指数:⭐️⭐️⭐️⭐️⭐️
时间:25.12.16
关键词:Tahoe-100M\药物扰动\通用细胞表示(universal cell embedding)
摘要:构建细胞预测模型需要系统性地绘制扰动如何重塑每个细胞的状态、功能和行为。本文我们推出Tahoe-100M——一个包含1亿个转录组谱的千兆级单细胞图谱,通过测量1100种小分子扰动对50种癌细胞系的影响,揭示其作用机制。我们的高通量Mosaic平台由高度多样化且平衡优化的“细胞村落”构成,能有效降低批次效应,并在单细胞分辨率下实现数千种条件的并行分析,其规模前所未有。作为迄今最大的单细胞数据集,Tahoe-100M使人工智能驱动模型能够学习情境依赖性功能,捕捉基因调控和网络动态的基本原理。虽然我们利用癌症模型和药物化合物构建该资源,但Tahoe-100M本质上是一个广泛适用的扰动图谱,支持对多种组织和情境下的细胞生物学进行更深入解析。通过公开发布该图谱,我们旨在加速系统生物学中稳健人工智能框架的创建与发展,最终提升我们在广泛应用中预测和调控细胞行为的能力。
心得:Tahoe-100M 提供的是「药物 × 基因 × 细胞」的三维张量。
你可以把「细胞系 + 药物 + 剂量」喂给训练好的模型,它就能输出「每个基因的表达变化」;把这些变化嵌入虚拟细胞(AIVC)的通用表示(UR),就能在电脑里「跑实验」、预测疗效、设计新组合,而不用再养细胞、加药、测序。
-
虚拟筛选:输入 1 000 种化合物,AIVC 可给出预期疗效排序。
-
患者数字孪生:把患者肿瘤细胞的突变谱输入,AIVC 模拟不同药物组合,挑出最有效的 3 种。
1.研究背景
-
早期研究把几百万个细胞混在一起测 RNA,只能得到“平均脸”。后来虽然单细胞技术成熟了,但一次实验通常只测几十种条件,数据量不够训练大模型。
-
只看细胞自然状态不够,CRISPR 敲基因、小分子加药等“扰动”手段来揭示因果逻辑。但现有扰动数据集规模小(10^4–10^5 细胞)、覆盖细胞系少、药物种类单一,导致 AI 模型容易过拟合,换个细胞系就失灵。
-
把多种细胞系混在一起做,节省试剂且消除批次差异。但传统做法每种细胞系单独培养、单独加药,96 孔板很快就被占满,成本高、批次噪音大。
-
过去的数据集要么细胞数少,要么药物种类单一,导致模型只能“死记硬背”。本文用规模+多样性双轮驱动,让 AI 真正“理解”药物机理。
-
过去缺少“标准化、大规模、多细胞系、多药物”的扰动基准,本文把数据统一嵌入 10 维 scVI 向量,形成通用细胞表示(UR),可直接对接虚拟细胞(AIVC)框架,可用于预测未见药物反应、模拟患者特异性突变背景下的疗效、指导下一步实验设计。
2.研究框架
每个Mosaic肿瘤由50个癌细胞系模型组成,这些模型被接种到96孔板中。每个孔接受药物扰动处理,24小时后,肿瘤被解离成单细胞,并使用Parse GigaLab试剂盒(这是一种通过组合条形码技术实现的可扩展单细胞RNA测序检测方法)进行条形码标记。这些条形码与已知的每个细胞系基因型相结合,从而实现治疗和细胞系的解卷积。
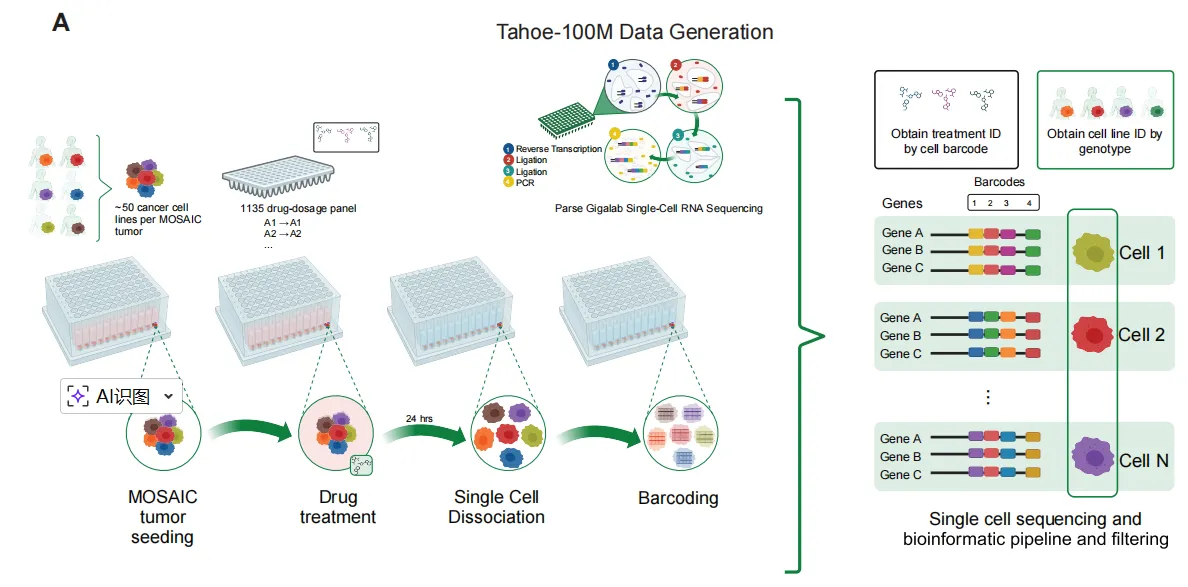
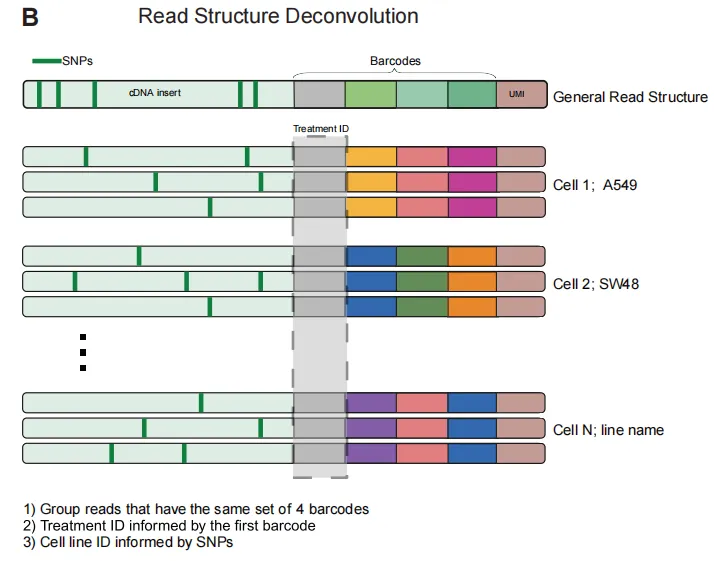
检测单细胞RNA测序文库中存在的遗传变异,并通过基于SNP的解卷积技术将细胞归类至其来源细胞系
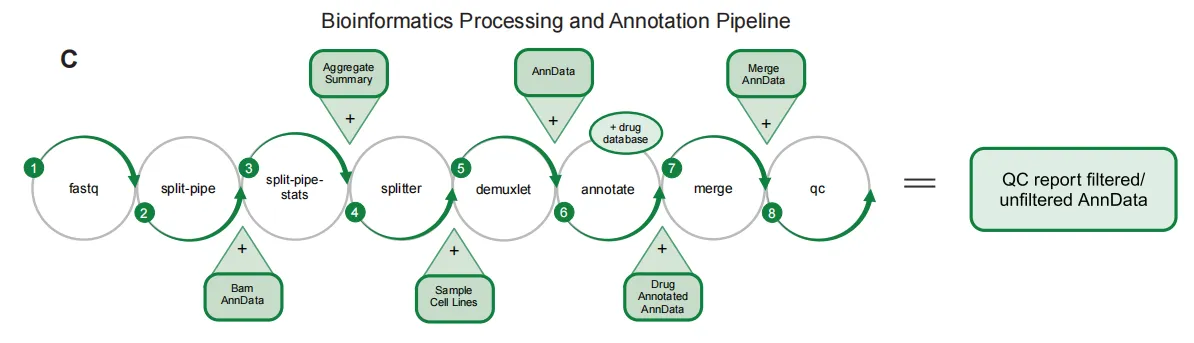
去卷积与生物信息学流程用于处理、注释和质量控制细胞,最终生成具有指定处理方式和细胞系身份的单细胞基因表达矩阵
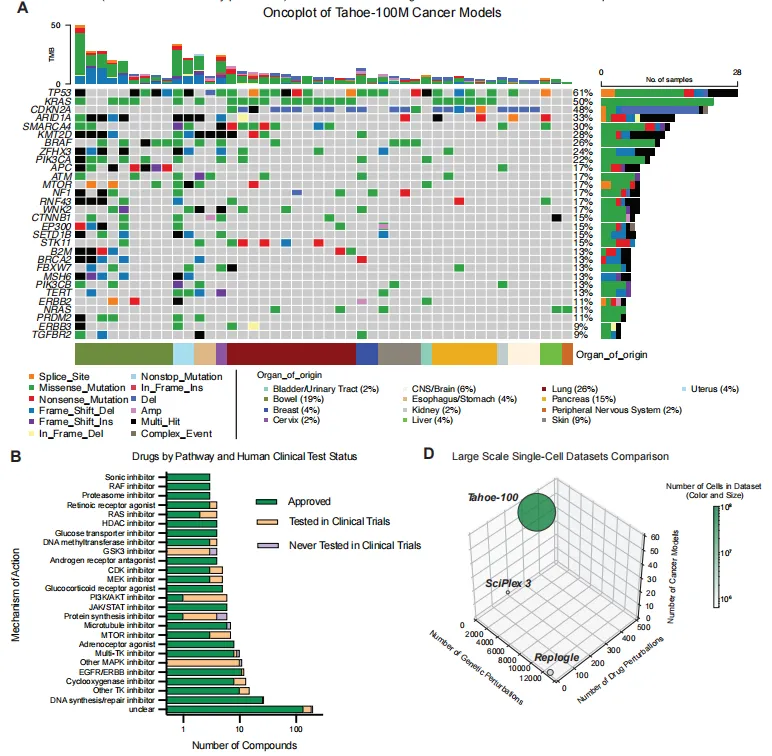
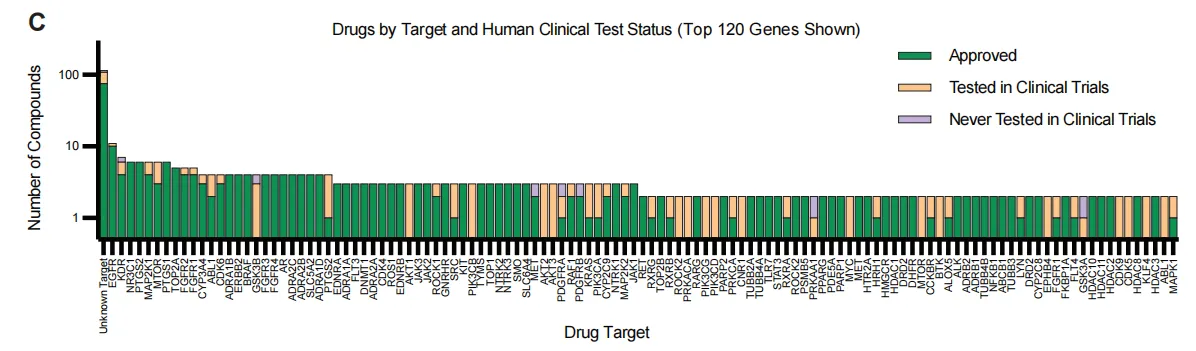
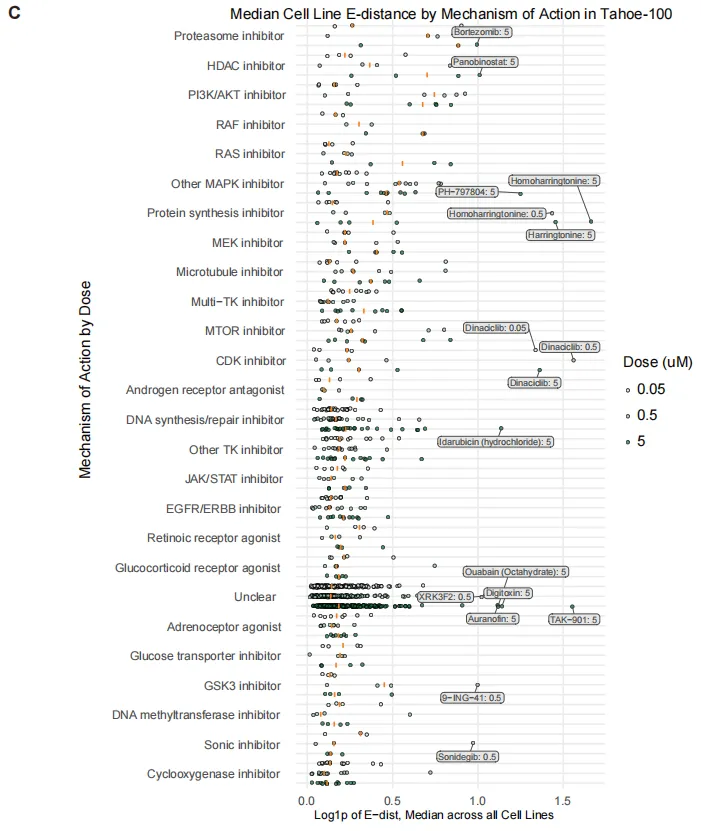
图2是对细胞系进行了药物扰动,这47个细胞系来源于13种不同器官(主要来自肺、肠道、胰腺和皮肤),携带多样化的驱动突变。该药物数据集共涵盖17,813种独特的细胞系-药物组合
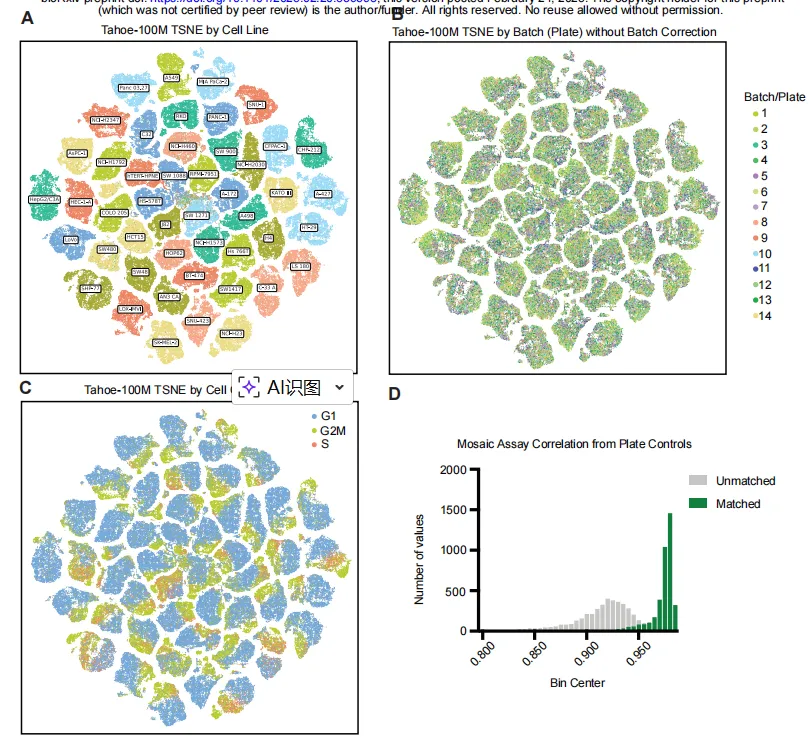
这个图3是先对细胞数据进行降维处理,为每个细胞生成10维嵌入向量。47个细胞系中分别随机挑选了14万个高质量细胞进行聚类展示,细胞在转录组空间中根据遗传特征(及细胞周期阶段)呈现明显分离,而非源自不同培养板,这表明约1亿个细胞的统一图谱中不存在显著的批次效应。A是癌症模型身份,B是细胞孔板,C是细胞周期,D是两个生物学重复96孔板之间基因表达向量相关性的皮尔逊相关系数分布。
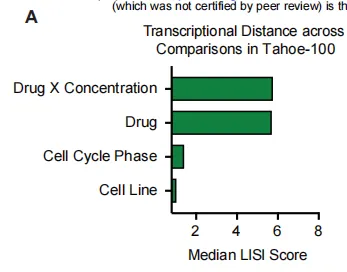
为了量化Tahoe-100M细胞群分组中的技术与生物学因素,我们基于数据子样本的不同元数据因子计算了局部逆辛普森指数(LISI)(Local Inverse Simpson Index,看一个细胞周围邻居“混不混”,判断批次效应强不强)。细胞系身份是最主要变异来源,药物剂量次之,批次效应最小。
利用scVI嵌入技术得到E-distance(扰动-对照距离,量化扰动群体与对照组的可分离性),随剂量增加而增大;使用 Sci-Plex3 及 CRISPRi 数据集也得到类似的效应。
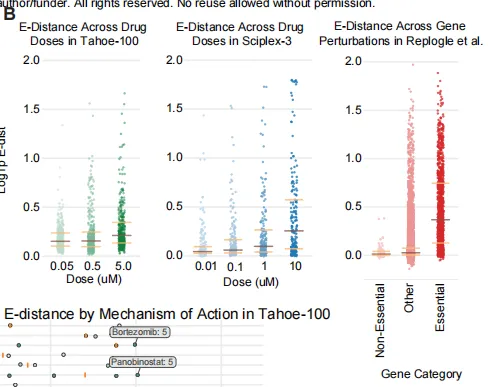
在按已报道的作用机制(MOA)对药物进行分层时,观察到若干显著的异常值。蛋白合成抑制剂(harringtonine)、CDK 抑制剂(dinaciclib)、HDAC/PI3K/蛋白酶体抑制剂等扰动最强。
差异基因集空间(图 5)
图 5. 差异基因集得分的降维可视化
-
A:把每个“细胞系-药物-剂量”伪 bulk 的 7 390 个 MSigDB 基因集得分做成矩阵。(对于基于差异表达基因集评分的降维)Atlas数据库中的单细胞数据点按共享的细胞模型和药物处理条件进行伪批量分组,计算7,390个基因集的差异基因表达评分。最终得到的61,927个细胞模型与药物处理伪批量组及7,390个基因集矩阵,作为两种不同降维可视化分析的输入数据。
-
B/C:t-SNE 与 NNMDS 着色按 E-distance → 高剂量药物落在图外围,低剂量靠近中心。
-
D/E:仅保留高 E-distance 的 MOA → HDAC、PI3K/AKT、蛋白酶体抑制剂等形成明显聚类。
-
F/G:KRAS-G12C 抑制剂 Adagrasib 与 pan-RAS 抑制剂 RMC-6236 在突变背景不同的细胞系中呈现清晰分离。 结论:基因集空间能区分不同药物机制与突变背景,支持 AI 预测。
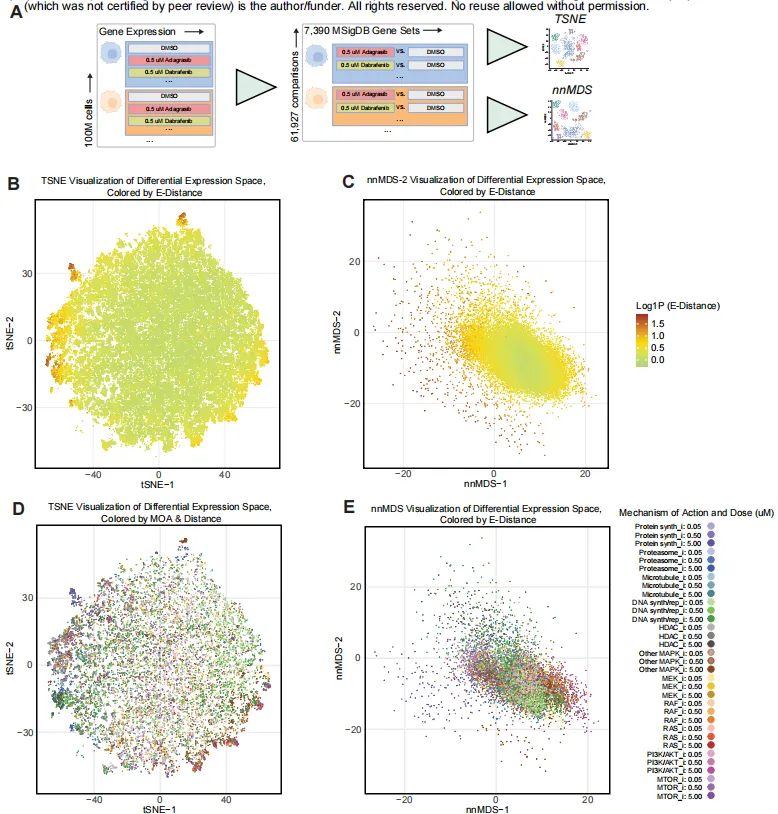
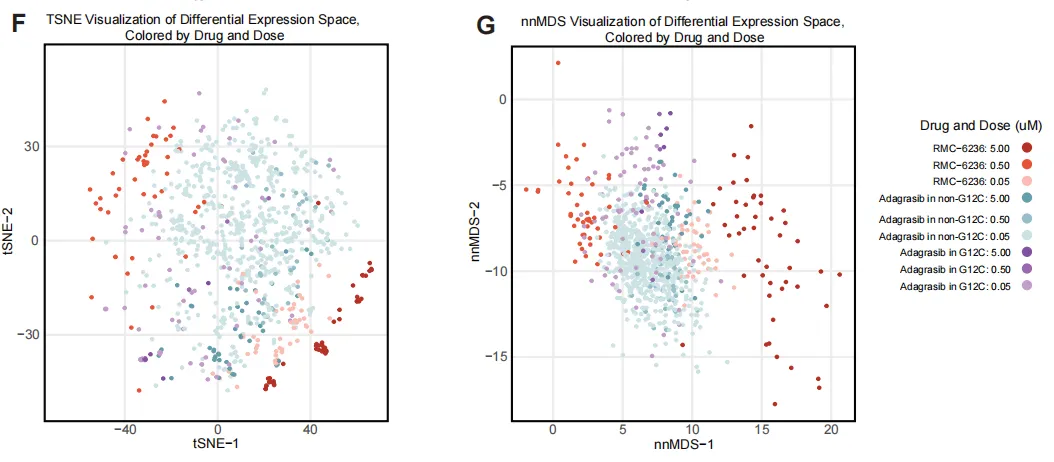
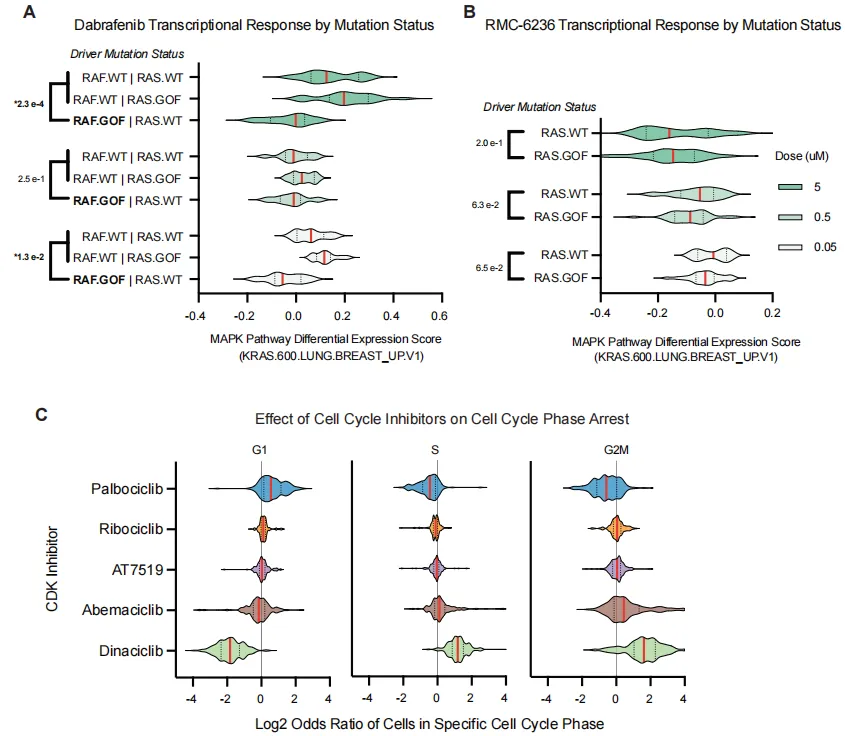
图 6. 选定药物的基因表达与细胞周期相位响应
-
A:Dabrafenib 像一把钥匙,只能插进 BRAF-V600E 这把“坏锁”,把信号通路关掉;对 KRAS 突变细胞无效,因为 KRAS 在 BRAF 上游,锁的位置不同。只有 BRAF-V600E 突变 的细胞在加药后通路得分显著下降(p = 0.00023)。KRAS 突变或野生型几乎没变化。
-
B:pan-RAS 抑制剂 RMC-6236 ,只有 KRAS 突变 细胞通路得分下降(最低剂量 *p = 0.063)
-
C:CDK 抑制剂 — Palbociclib 诱导 G1 阻滞,Dinaciclib 诱导 G2/M 阻滞
-
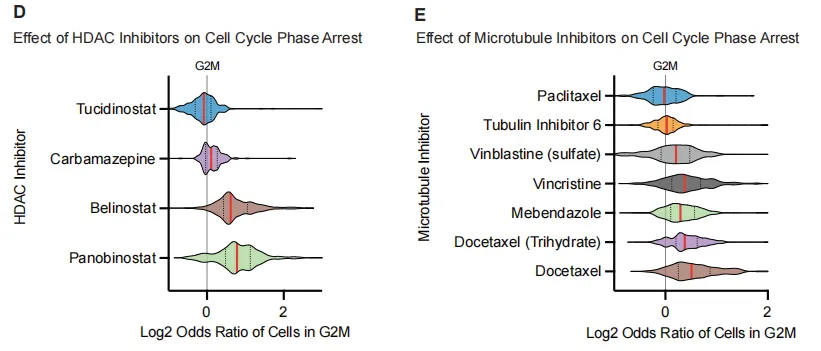
D/E:HDAC 与微管抑制剂普遍引起 G2/M 阻滞,但不同分子间存在差异(如 Tucidinostat 反而减少 G2/M)。 结论:单细胞分辨率能揭示突变背景与药物机制如何共同决定细胞命运。
讨论
-
Tahoe-100M 把数据量、细胞系多样性、药物-剂量组合同时放大 1–2 个数量级,满足 AI 对“见多识广”的需求。
-
即使在遗传背景完全相同的细胞系里,也存在多个转录亚群,对药物敏感度不同。单细胞分辨率首次把这些亚群拆开,避免 bulk 实验“平均脸”陷阱。这种异质性正是肿瘤耐药的根源,也是 AI 模型必须学习的“噪声”。
-
用 RAS/RAF 通路签名证明:Dabrafenib 只在 BRAF-V600E 突变细胞中有效,RMC-6236 只在 KRAS 突变细胞中有效。说明单细胞转录指纹可替代部分湿实验,用于预测“谁会对药有反应”。为精准肿瘤学提供“突变-药物”匹配表,减少盲目试药。
-
CDK、HDAC、微管抑制剂分别把细胞阻滞在 G1 或 G2/M,与它们的已知靶点一致。但不同分子即使同属一类,也可能出现差异(如 Tucidinostat 反而减少 G2/M)。提示单细胞数据可用来细分药物机制,发现新的生物标志物。
-
公开下载:2025-02-25 起通过 Arc Virtual Cell Atlas 免费提供。期待社区用它训练下一代虚拟细胞(AIVC),实现“干-湿闭环”实验设计。下一步:扩展至原代细胞、3D 类器官、体内模型,最终走向患者数字孪生。
一些重要名词
demuxlet:用细胞自带的 DNA“指纹”(SNP)把混在一起的单细胞重新认领回原来的细胞系。Mosaic 平台把 50 种癌细胞混在一个孔里,测序后靠 demuxlet 把每个细胞准确分到“它原来属于哪个细胞系”
pseudobulk’ UMI counts:把很多单细胞的 UMI(唯一分子标签)计数加在一起,假装成一个“大样本”的表达量。用来比较“同一细胞系-药物”在两块 96 孔板(生物学重复)之间的相关性,证明实验可重复(图 3D)。
MOA:把 379 种药物分成 25 类 MOA(如 CDK 抑制剂、HDAC 抑制剂),用来比较不同机制引起的转录变化(图 4C、5D-E)。
MedChemExpress:一个商业化合物库网站,提供药物的化学结构、靶点、临床状态等信息。作者用爬虫+GPT-4o 从 MCE 抓取 379 种药物的靶点基因和作用机制,快速建立“药物-靶点”映射表,省去人工查文献。提取了化合物描述和靶点列表(如已知)的字符串
scVI model:一种深度学习模型,把成千上万个基因的表达压缩成 10 个数字(latent vector),同时去掉技术噪音。
-
训练集:89 M 细胞 × 62 k 基因 → 10 维向量。
-
用途:下游做 E-distance、细胞周期分析、可视化(图 3-6)都基于这 10 个数,既省算力又降噪。